



MoleculeModify
MoleculeModify[mol,"mod"]
gives a molecule or list of molecules derived from the molecule mol by applying the modification "mod".
MoleculeModify[mol,{"mod",specs}]
gives a molecule or list of molecules derived from the molecule mol by applying the modification "mod" with additional specifications specs.
MoleculeModify["mod"]
represents an operator form of MoleculeModify that can be applied to a molecule.
Details and Options
- Operations that modify the molecule's atom list:
-
{"AddAtom",atom} add a disconnected atom {"AddAtom",{atom1,atom2,…}} add a list of atoms {"DeleteAtom",a} delete the atom with index a {"DeleteAtom",{a1,a2,…}} delete the indicated atoms {"ReplaceAtom",aatom} replace the atom with index a with atom {"ReplaceAtom",{a1atom1,…}} replace atoms ai with atomi - atom should have one of the following forms:
-
"sym" atom with atomic symbol "sym" (e.g. "C", "Cl") Entity["Element",element] atom of a given element Entity["Isotope",isotope] atom of the specified isotope Atom["sym","prop"val,…] atom with specified properties - "AddAtom" will add a disconnected atom, with no bonds to the rest of the molecule. To add a new atom and bond together use "AddBond".
- "DeleteAtom" will remove any bonds to the atom in question.
- When removing or replacing an atom, the number of explicit hydrogen atoms may be adjusted to maintain proper valence. Disconnected hydrogen atoms will be removed. To disable this behavior, use the option ValenceErrorHandlingFalse.
- Implicit hydrogen atoms are added to the added atoms as normal. To disable implicit hydrogens, use "HydrogenCount"None in the new atom specification.
- Operations that modify atom properties:
-
{"SetFormalCharge",aval} set the formal charge of the atom with index a to val {"SetMassNumber",aval} set the mass number of the atom with index a to val {"SetUnpairedElectronCount",aval} set the number of radical electrons for the given atom - To change a property for multiple atoms, use a list of rules {a1val1,a2val2,…}.
- Operations that modify the molecule's bond list:
-
{"AddBond",{a1,a2}} add a single bond between atoms with indices a1 and a2 {"AddBond",Bond[{a1,a2},"type"]} add a bond of the given type between the given atoms {"AddBond",Bond[{a1,atom},type]} a bond between an atom with index a1 and a new atom represented by atom {"DeleteBond",{a1,a2}} remove the bond between the atoms with the given indices {"SetBondType",{a1,a2}"type"} set the bond type for the bond between atoms with indices a1 and a2 -
"SetAromaticity" convert localized single and double bonds to aromatic "Kekulize" convert aromatic bonds to localized single and double bonds {"RenumberAtoms",{a1,a2,…}} reorder the atoms list so that the atom with index ai appears at position i in the atoms list {"SetMetaInformation",meta} set the molecule's MetaInformation to meta, which should be an association or key-value pair "MakeHydrogensExplicit" convert any implicit hydrogens to explicit "MakeHydrogensImplicit" remove explicit hydrogen atoms when their presence can be inferred by normal valence rules - Information about a molecule's 3D arrangement, independent of the 3D geometry, is stored in the StereochemistryElements option value. The following operations modify the stereo elements:
-
"AssignStereochemistryFromCoordinates" assign tetrahedral and double bond stereochemistry elements from 3D coordinates "RemoveStereochemistry" remove any defined stereochemistry - To change the defined stereochemistry for an atom or bond, use one of the following operations:
-
{"SetAtomChirality",aval} set the chiral value for an atom to "R" or "S" {"SetBondStereo",{a1,a2}val} set the bond stereo value to either "E" or "Z" - Changing the stereo for an atom or bond will affect the 3D coordinates that are generated on demand. If the molecule contains 3D coordinates already, these are discarded when changing the stereochemistry.
-
{"DeleteSubstructure",patt} delete all occurrences of patt in the molecule {"ReplaceSubstructure",pattrepl} replace all occurrences of patt with repl {"ReplaceSubstructure",pattrepl,attchmts} replace patt with repl, using the specified attachment points between pattern and replacement {"ReplaceSubstructureList",pattrepl} replace each occurrence of patt with repl and return a list - Here repl should be a Molecule object or SMILES string, and patt can have one of the following forms:
-
MoleculePattern[…] a valid molecule pattern object {a1,a2,…} a list of atom indices constituting a connected substructure {{a1,a2,…},{b1,b2,…},…} a list of lists of atom indices, each of which will be replaced with repl - attchmts should be a list of rules {p1r1,…} from pattern atom index to replacement atom index. If no attachment points are provided, the first and last atoms from the pattern and replacement are matched.
- When a molecule is plotted in 3D or 2D, coordinates are taken from the AtomCoordinates and AtomDiagramCoordinates option values, respectively. If no coordinates are present in the molecule expression, they are generated automatically.
- The following operations will precompute the automatic coordinates and return a Molecule object containing them:
-
"ComputeAtomCoordinates" compute automatic 3D coordinates for the molecule {"ComputeAtomCoordinates",opts} compute atom coordinates using the options opts {"GenerateConformers",n} compute 3D coordinates n times with a different seed value and return a list of molecules "ComputeAtomDiagramCoordinates" compute 2D coordinates - "ComputeAtomCoordinates" will make all implicit hydrogens explicit in order to embed the molecule.
- "ComputeAtomCoordinates" takes the following options:
-
"Canonicalize" True whether to canonicalize the coordinates "EnergyMinimize" True whether to run a force-field minimization on the coordinates RandomSeeding 1234 how to seed randomness - "GenerateConformers" will return unminimized conformers by default.
- "ComputeAtomDiagramCoordinates" will make implicit those hydrogens attached to carbon, and make explicit those attached to a heteroatom, to match the default output of MoleculePlot. Use {"ComputeAtomDiagramCoordinates",IncludeHydrogensTrue} to include all atoms.
- To change the coordinates for a single atom, use:
-
{"SetAtomPosition",a{x,y,z}} set the position of the specified atom - To canonicalize atom coordinates without changing internal geometry, use:
-
"CanonicalizeAtomCoordinates" translate the molecule's center of mass to the origin and align the principal axes with the Cartesian axes - To invert the local geometry around a particular atom, use:
-
{"InvertAroundAtom",a} invert the geometry around atom with index 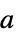
{"InvertAroundAtom",{a1,a2,…}} invert the geometry around several atoms - Tetrahedral atoms are inverted by exchanging the two smallest movable ligands. Trigonal pyramidal atoms are inverted by moving the smallest movable ligand. Movable atoms are either not in rings or are in the same ring. The coordinates of the moving atoms are transformed with a twofold rotation, thus preserving all other stereocenters in the molecule.
- For general geometry transformations use the following:
-
{"TransformAtomCoordinates",tfun} apply the transformation function tfun to the coordinates - Here tfun should be a TransformationFunction object, e.g. TranslationTransform, ReflectionTransform, etc., or any arbitrary function that operates on and returns a QuantityArray object.
- The following operations modify the internal coordinates:
-
{"SetBondLength", {a1,a2}val} set the bond length between the specified atoms {"SetBondAngle", {a1,a2,a3}val} set the angle between the specified atoms {"SetTorsionAngle",{a1,a2,a3,a4}val} set the torsion angle for a group of four consecutively bonded atoms - These operations also support a list of length or angle changes, e.g. {{a1,a2}val1,…}.
- Here val should be a Quantity object or numeric. If numeric, the units are taken to be "Angstroms" or "AngularDegrees".
- For "SetBondLength", atoms a1 and a2 must be bonded and the bond between them should not be in a ring. All atoms connected to atom a2 are moved.
- For "SetBondAngle", atoms {a1,a2} and {a2,a3} should be bonded and both bonds should not be in rings. All atoms connected to atom a3 are moved.
- For "SetTorsionAngle", atoms {a1,a2}, {a2,a3} and {a3,a4} should be bonded, and bond {a2,a3} should not be in a ring. All atoms connected to atom a4 are moved.
- To minimize the coordinates with respect to the MMFF force field, use the following:
-
"EnergyMinimizeAtomCoordinates" minimize the total force-field energy {"EnergyMinimizeAtomCoordinates",ff} minimize the energy using the force field ff, one of "UFF", "MMFF94" or "MMFF94s" {"EnergyMinimizeAtomCoordinates",constraints} minimize the energy with the given constraints added to the force field - constraints should be a list of bond, angle or torsion constraints of the following forms:
-
{a1,a2}d specify a distance between two atoms {a1,a2,a3}θ specify an angle among three atoms {a1,a2,a3,a4}θ specify a torsion angle among four atoms <…> an association with keys "Atoms", "Value" and "ForceConstant"
Atom Modifications
Bond Modifications
General Modifications
Stereochemistry Modifications
Substructure Deletion and Replacement
Generating Coordinates
Geometry Modifications
Examples
open all close allBasic Examples (4)
MoleculeModify[Molecule["propane"], {"AddAtom", "C"}]MoleculeModify[%, {"AddBond", Bond[{2, 12}]}]ToEntity[%]Add a bond to turn hexane into cyclohexane:
m = Molecule["hexane"]MoleculeModify[m, {"AddBond", Bond[{1, 6}, "Single"]}]Delete a bond to convert cyclohexane to hexane:
m = Molecule["cyclohexane"]MoleculeModify[m, {"DeleteBond", Bond[{4, 3}, "Single"]}]Replace all nitro groups in TNT with methyl groups:
nitro = MoleculePattern["[#7](~[#8])~[#8]"];MoleculeModify[Molecule["trinitrotoluene"], {"ReplaceSubstructure", nitro -> "C"}]Use "ReplaceSubstructureList" to replace one nitro group at a time:
MoleculeModify[Molecule["trinitrotoluene"], {"ReplaceSubstructureList", nitro -> "C"}]Scope (41)
"AddAtom" (1)
MoleculeModify[Molecule[{Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"],
Atom["C"], Atom["C"], Atom["H"], Atom["H"], Atom["H"], Atom["H"], Atom["H"], Atom["H"],
Atom["Cl"], Atom["Cl"]}, {Bond[{1, 2}, "Aromatic"], Bond ... romatic"], Bond[{10, 5}, "Aromatic"],
Bond[{3, 11}, "Single"], Bond[{4, 12}, "Single"], Bond[{6, 13}, "Single"],
Bond[{7, 14}, "Single"], Bond[{8, 15}, "Single"], Bond[{9, 16}, "Single"],
Bond[{17, 1}, "Single"], Bond[{18, 2}, "Single"]}], {"AddAtom", "Fe"}]MoleculeModify[Molecule[{Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"],
Atom["C"], Atom["C"], Atom["H"], Atom["H"], Atom["H"], Atom["H"], Atom["H"], Atom["H"],
Atom["Cl"], Atom["Cl"]}, {Bond[{1, 2}, "Aromatic"], Bond ... romatic"], Bond[{10, 5}, "Aromatic"],
Bond[{3, 11}, "Single"], Bond[{4, 12}, "Single"], Bond[{6, 13}, "Single"],
Bond[{7, 14}, "Single"], Bond[{8, 15}, "Single"], Bond[{9, 16}, "Single"],
Bond[{17, 1}, "Single"], Bond[{18, 2}, "Single"]}], {"AddAtom", {"Na+", "Cl-"}}]"AddBond" (1)
"AssignStereochemistryFromCoordinates" (1)
Use a molecule's 3D coordinates to infer stereochemistry. First create a molecule with atoms, bonds and coordinates, but no defined stereochemistry:
m = Molecule[IconizedObject[«atom list»], IconizedObject[«bond list»], AtomCoordinates -> QuantityArray[StructuredArray`StructuredData[{47, 3},
{{{2.4265, -1.6857, -1.6031}, {3.9689, 0.2041, 0.0446}, {2.2747, 1.303, 2.1955},
{-1.0178, -1.3778, 0.9927}, {-4.3652, -2.3641, 0.1309}, {-2.5758, -3.9668, 1.4015},
{1.3208, -1.9591 ... 692, 0.7349, 1.5371}, {0.7256, 1.4585, -2.1836}, {-3.2418, 4.7619, -0.8261},
{-1.913, 5.208, -1.8817}, {2.3128, -3.6443, -2.7289}, {4.2015, 1.6396, -1.6863},
{1.5146, 3.3749, 1.7047}, {0.4457, 1.1452, 3.5146}}, "Angstroms", {{1}, {2}}}]]]m2 = MoleculeModify[m, "AssignStereochemistryFromCoordinates"]m2["StereochemistryElements"]//Dataset"CanonicalizeAtomCoordinates" (1)
Import a structure from an "XYZ" file and canonicalize the 3D coordinates:
m = Import["ExampleData/caffeine.xyz"]After importing, compute the molecule's center of mass and principal axes:
MoleculeValue[m, {"CenterOfMass", "PrincipalAxes"}]"CanonicalizeAtomCoordinates" will translate the molecule, move the center of mass to the origin and rotate to align the principal axes with the Cartesian axes:
m2 = MoleculeModify[m, "CanonicalizeAtomCoordinates"];
MoleculeValue[m2, {"CenterOfMass", "PrincipalAxes"}]"ComputeAtomCoordinates" (2)
When a molecule is created from an identifier like a SMILES string or name, coordinates are not computed immediately:
m = Molecule["FCCN1CCC(CC1)COc1ccc(cc1)C#N"];
Options[m]Use "ComputeAtomCoordinates" to compute the coordinates and store them in the molecule expression:
m2 = MoleculeModify[m, "ComputeAtomCoordinates"];
Options[m2]Generate different conformations using the RandomSeeding option:
{m1, m2, m3} = MoleculeModify[Molecule["4-[[1-(2-fluoroethyl)piperidin-4-yl]methoxy]benzonitrile"], {"ComputeAtomCoordinates", RandomSeeding -> #}]& /@ {1, 12, 23};MoleculePlot3D /@ {m1, m2, m3}The resulting molecules have different conformational energy:
MoleculeValue[{m1, m2, m3}, "MMFFEnergy"]"ComputeAtomDiagramCoordinates" (2)
When a molecule is visualized in 2D using MoleculePlot, hydrogen atoms attached to carbon atoms are hidden by default. Only the hydroxyl hydrogen is shown by default in octanol:
MoleculePlot[Molecule["2-octanol"]]"ComputeAtomDiagramCoordinates" will remove those hydrogens that are not shown, and make explicit those that are shown:
mol = MoleculeModify[Molecule["2-octanol"], "ComputeAtomDiagramCoordinates"]The coordinates are now stored in the molecule expression:
Options[mol]As there is only one hydrogen shown by default, the returned molecule has one explicit hydrogen:
AtomList[mol, "H", "AtomIndex", IncludeHydrogens -> "ExplicitOnly"]The coordinates match exactly those used in a molecule plot:
coords = mol["AtomDiagramCoordinates", IncludeHydrogens -> "ExplicitOnly"];Show[Graphics[{Yellow, Disk[#, .2]& /@ coords}], MoleculePlot[mol]]Compute the atom diagram coordinates showing the default hydrogens, all hydrogens or none:
highlightCoordinates[option_] := Module[{mol}, mol = MoleculeModify[Molecule["2-octanol"], {"ComputeAtomDiagramCoordinates", IncludeHydrogens -> option}];
Show[
Graphics[{Yellow, Disk[#, .2]& /@ mol["AtomDiagramCoordinates", IncludeHydrogens -> "ExplicitOnly"]}], MoleculePlot[mol, IncludeHydrogens -> option]]
]GraphicsColumn[highlightCoordinates /@ {Automatic, True, False}]"DeleteAtom" (2)
Convert hexanol to hexane using "DeleteAtom":
m = Molecule["2-hexanol"]AtomList[m, "O", "AtomIndex"]MoleculeModify[m, {"DeleteAtom", %}]Find an Entity to match:
ToEntity[%]When an atom is deleted, any hydrogens attached to it are also removed, and unfilled valences on heavy atoms are filled by implicit hydrogens. Use ValenceErrorHandling->False to disable this behavior and leave unpaired electrons instead:
MoleculeModify[Molecule@"CCCP[C@@H]1C[C@@H]1C", {"DeleteAtom", 4}, ValenceErrorHandling -> #]& /@ {True, False}MoleculePlot /@ %"DeleteBond" (2)
Delete an acyclic bond to create disconnected structures:
m = Molecule["diphenylamine"]BondList[m, Bond[{"N", "C"}]]MoleculeModify[m, {"DeleteBond", First@%}]MoleculePlot@%MoleculeModify[Molecule[{"C", "C", "C", "N", "C", "C", "C", "N", "O", "S", "O"},
{Bond[{4, 5}, "Single"], Bond[{5, 3}, "Aromatic"], Bond[{3, 2}, "Aromatic"],
Bond[{2, 1}, "Aromatic"], Bond[{1, 7}, "Aromatic"], Bond[{7, 6}, "Aromatic"],
Bond[{1, 10}, "Single"], Bond[{10, 9}, "Double"], Bond[{10, 11}, "Double"],
Bond[{10, 8}, "Single"], Bond[{6, 5}, "Aromatic"]}, {}], {"DeleteBond", {7, 6}}]"DeleteSubstructure" (1)
This molecule has two amine groups:
MoleculePlot[mol = Molecule[{"C", "C", "C", "N", "C", "C", "C", "N", "O", "S", "O"},
{Bond[{4, 5}, "Single"], Bond[{5, 3}, "Aromatic"], Bond[{3, 2}, "Aromatic"],
Bond[{2, 1}, "Aromatic"], Bond[{1, 7}, "Aromatic"], Bond[{7, 6}, "Aromatic"],
Bond[{1, 10}, "Single"], Bond[{10, 9}, "Double"], Bond[{10, 11}, "Double"],
Bond[{10, 8}, "Single"], Bond[{6, 5}, "Aromatic"]}, {}]]MoleculeModify[mol, {"DeleteSubstructure", MoleculePattern["[NH2]"]}]MoleculePlot[%]"EnergyMinimizeAtomCoordinates" (1)
Import a structure from an "XYZ" file and use the MMFF force field to minimize the energy:
m = Import["ExampleData/caffeine.xyz"]m2 = MoleculeModify[m, "EnergyMinimizeAtomCoordinates"]Verify that the conformational energy has lowered:
MoleculeValue[{m, m2}, "MMFFEnergy"]Show the new structure together with the old:
Show[
MoleculePlot3D[m, ColorRules -> {_ -> Directive[Opacity[0.4], Gray]}], MoleculePlot3D[m2]
]"GenerateConformers" (1)
It is often desirable to have a set of conformations for a molecule, for example, to sample a larger portion of the energy landscape. Generate 100 conformers for cyclohexane:
mols = MoleculeModify[Molecule["cyclohexane"], {"GenerateConformers", 100}];The conformers will have a large energy spread:
Histogram[MoleculeValue[mols, "MMFFEnergy"], PlotTheme -> "Detailed"]After minimizing the conformations using a force field, they fall into two groups energetically:
mols2 = MoleculeModify["EnergyMinimizeAtomCoordinates"] /@ mols;
Histogram[MoleculeValue[mols2, "MMFFEnergy"], PlotTheme -> "Detailed"]These groupings correspond to the so-called "chair" and "twist-boat" conformations, which are global and local minima on the potential energy surface, respectively. Select one conformer from each group and plot in 3D:
twistBoat = SelectFirst[mols2, #["MMFFEnergy"] > Quantity[0, "KilocaloriesThermochemical"/"Moles"]&];
chair = SelectFirst[mols2, #["MMFFEnergy"] < Quantity[0, "KilocaloriesThermochemical"/"Moles"]&];GraphicsRow[MoleculePlot3D[#, ViewPoint -> Left, PlotTheme -> "Tubes"]& /@ {chair, twistBoat}, ImageSize -> 500]"InvertAroundAtom" (3)
mol = Molecule["R-sec-butanol"];
MoleculePlot3D[%, IconizedObject[«viewOptions»]]invMol = MoleculeModify[mol, {"InvertAroundAtom", 3}];
MoleculePlot3D[%, IconizedObject[«viewOptions»]]MoleculeValue[{mol, invMol}, {"AtomChirality", 3}]Atoms in rings can also be inverted. Invert one of the carbinol atoms of cis-cyclohexan-1,3-diol:
mol = Molecule["cis-cyclohexan-1,3-diol"];
MoleculePlot3D[%, {2}, IconizedObject[«viewOptions»]]invMol = MoleculeModify[mol, {"InvertAroundAtom", 2}];
MoleculePlot3D[%, {2}, IconizedObject[«viewOptions»]]MoleculeValue[{mol, invMol}, {"AtomChirality", 2}]The configuration at the other carbinol has not changed:
MoleculeValue[{mol, invMol}, {"AtomChirality", 6}]Several atoms can be inverted at once. Show (-)-menthol:
minusMenthol = Molecule["O[C@H]1[C@H](C(C)C)CC[C@@H](C)C1"];
MoleculePlot3D[%, {{2, 9}}, IconizedObject[«viewOptions»]]Invert atoms 2 (bearing the hydroxyl group) and 9 (bearing the methyl group) to make (+)-neoisomenthol:
plusNeoisomenthol = MoleculeModify[minusMenthol, {"InvertAroundAtom", {2, 9}}];
MoleculePlot3D[%, {{2, 9}}, IconizedObject[«viewOptions»]]MoleculeValue[minusMenthol, {"AtomChirality", {2, 3, 9}}]MoleculeValue[plusNeoisomenthol, {"AtomChirality", {2, 3, 9}}]"Kekulize" (1)
"MakeHydrogensImplicit" (1)
Remove all the hydrogen atoms from a molecule:
mol = Molecule["cholesterol"]mol["ElementCounts", IncludeHydrogens -> "ExplicitOnly"]mol2 = MoleculeModify[mol, "MakeHydrogensImplicit"]mol2["ElementCounts", IncludeHydrogens -> "ExplicitOnly"]"RemoveStereochemistry" (1)
Remove explicit stereo information from a molecule, leaving the chirality undefined:
mol = Molecule["L-threonine"]AtomList[mol, Atom["AtomChirality" -> Except[None]], {"AtomIndex", "AtomChirality"}]mol2 = MoleculeModify[mol, "RemoveStereochemistry"]AtomList[mol2, Atom["AtomChirality" -> Except[None]], {"AtomIndex", "AtomChirality"}]"RenumberAtoms" (1)
"ReplaceAtom" (1)
"ReplaceSubstructure" (1)
Replace the hydroxyl in the aliphatic D ring of estradiol with a carbonyl to make estrone:
estradiol = Molecule["C[C@]12CC[C@@H]3c4ccc(cc4CC[C@H]3[C@@H]1CC[C@@H]2O)O"]estrone = MoleculeModify[estradiol, {"ReplaceSubstructure", MoleculePattern["CO"] -> "C=O"}]MoleculePlot /@ {estradiol, estrone}"ReplaceSubstructureList" (1)
"SetAromaticity" (1)
Change localized single and double bonds to aromatic:
m = Molecule[{Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["H"], Atom["H"],
Atom["H"], Atom["H"], Atom["H"], Atom["H"]}, {Bond[{1, 2}, "Double"], Bond[{2, 3}, "Single"],
Bond[{3, 4}, "Double"], Bond[{4, 5}, "Single"], Bond[{5, 6}, "Double"], Bond[{6, 1}, "Single"],
Bond[{1, 7}, "Single"], Bond[{2, 8}, "Single"], Bond[{3, 9}, "Single"], Bond[{4, 10}, "Single"],
Bond[{5, 11}, "Single"], Bond[{6, 12}, "Single"]}, IncludeAromaticBonds -> False];BondList[m, Bond[{"C", "C"}]]m2 = MoleculeModify[m, "SetAromaticity"]BondList[m2, Bond[{"C", "C"}]]"SetAtomChirality" (1)
Convert D-glucose to D-mannose:
glucose = Molecule["D-glucose"]AtomList[glucose, Atom["AtomChirality" -> Except[None]], {"AtomIndex", "AtomChirality"}]mannose = MoleculeModify[glucose, {"SetAtomChirality", 3 -> "S"}]AtomList[mannose, Atom["AtomChirality" -> Except[None]], {"AtomIndex", "AtomChirality"}]MoleculePlot3D[#, Style[3, Blue], ImageSize -> Small]& /@ {glucose, mannose}//Row"SetAtomPosition" (1)
Import a molecule and set the coordinate for a single atom:
m = Import["ExampleData/caffeine.xyz"];
m2 = MoleculeModify[m, {"SetAtomPosition", 2 -> {Quantity[1, "Angstroms"], Quantity[2, "Angstroms"], Quantity[3, "Angstroms"]}}]Normal@MoleculeValue[m2, {"AtomCoordinates", 2}]"SetBondAngle" (3)
Set the angle between three atoms:
m = Molecule["acetone"]MoleculeModify[m, {"SetBondAngle", {1, 2, 4} -> Quantity[120, "AngularDegrees"]}]%[{"BondAngle", {1, 2, 4}}]MoleculeModify[m, {"SetBondAngle", {{1, 2, 4} -> Quantity[(3*Pi)/4, "Radians"], {1, 2, 3} -> Quantity[106., "AngularDegrees"]}}]%[{"BondAngle", {{1, 2, 4}, {1, 2, 3}}}]Angles cannot be set for nonbonded atoms:
MoleculeModify[Molecule[{Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["N"], Atom["C"], Atom["O"], Atom["C"],
Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["H"],
Atom["H"], Atom["H"], Atom["H"], Atom["H"], Atom["H"], A ... "], Bond[{13, 34}, "Single"], Bond[{13, 35}, "Single"],
Bond[{13, 36}, "Single"], Bond[{14, 37}, "Single"], Bond[{14, 38}, "Single"],
Bond[{14, 39}, "Single"], Bond[{15, 40}, "Single"], Bond[{15, 41}, "Single"],
Bond[{15, 42}, "Single"]}], {"SetBondAngle", {9, 6, 7} -> Quantity[103, "AngularDegrees"]}]Bond angles in a ring cannot be set:
MoleculeModify[Molecule["benzene"], {"SetBondAngle", {1, 2, 3} -> Quantity[115, "AngularDegrees"]}]"SetBondLength" (1)
Set the distance between two bonded atoms:
mol = Molecule["chloroform"]mol[{"BondLength", {2, 5}}]MoleculeModify[mol, {"SetBondLength", {2, 5} -> Quantity[1.3, "Angstroms"]}]%[{"BondLength", {2, 5}}]MoleculeModify[mol, {"SetBondLength", {1, 3} -> Quantity[1.3, "Angstroms"]}]"SetBondStereo" (1)
Convert natural E-cinnamaldehyde to the Z-form:
cinnE = Molecule["c1ccc(cc1)/C=C/C=O"]cinnZ = MoleculeModify[cinnE, {"SetBondStereo", {7, 8} -> "Z"}]//MoleculeModify["ComputeAtomCoordinates"]MoleculePlot3D[#, ImageSize -> Small]& /@ {cinnE, cinnZ}//Row"SetBondType" (1)
"SetFormalCharge" (1)
"SetMassNumber" (1)
Label the C-2 of mevalonolactone with 13C by setting the mass number to 13:
mvl = Molecule["(4R)-4-hydroxy-4-methyloxan-2-one"]MoleculeModify[mvl, {"SetMassNumber", 9 -> 13}]Use AtomList to find the labeled carbon:
AtomList[%, Atom["MassNumber" -> 13], "AtomIndex"]Label the 5-pro-S hydrogen of mevalonolactone with deuterium by setting the mass number to 2:
labeledH = MoleculeModify[mvl, {"SetMassNumber", 19 -> 2}]MoleculePlot3D[labeledH, Atom["MassNumber" -> 2]]"SetMetaInformation" (1)
Often, laboratories use a compound numbering system for samples and notebooks, and these identification numbers are used by chemists when writing internal reports, research papers and patents. Use the molecule's meta-information to store this kind of data.
mol = MoleculeModify[Molecule[{Atom["O"], Atom["C"], Atom["C"], Atom["C"], Atom["O"], Atom["C"], Atom["C"], Atom["O"],
Atom["C"], Atom["C"], Atom["C"], Atom["O"], Atom["C"], Atom["C"], Atom["C"], Atom["C"],
Atom["Cl"], Atom["C"], Atom["O"], Atom["C"], Atom["C"], ... r" -> 6,
"Direction" -> "Counterclockwise", "FiducialAtom" -> 5, "Ligands" -> {7, 14, 2}],
Association["StereoType" -> "Tetrahedral", "ChiralCenter" -> 14, "Direction" -> "Clockwise",
"FiducialAtom" -> 13, "Ligands" -> {15, 6, 31}]}], {"SetMetaInformation", <|"C-number" -> "123456-789"|>}]mol["MetaInformation"]Store experimental data in this property:
mol = MoleculeModify[mol, {"SetMetaInformation", <|"SpecificRotation" -> {Quantity[364, "AngularDegrees"], "@ 20°C, sat. CHCl3"}|>}]mol["MetaInformation"]Use an empty association to remove the metadata:
MoleculeModify[mol, {"SetMetaInformation", <||>}]%["MetaInformation"]"SetTorsionAngle" (1)
Set the geometry of a molecule to a desired conformation, for example, for an internal hydrogen bond:
mol = Molecule["OCCC=O", IncludeHydrogens -> True];mol2 = MoleculeModify[mol, {"SetTorsionAngle", {{6, 1, 2, 3} -> 60, {1, 2, 3, 4} -> -60, {2, 3, 4, 5} -> 60}}];MoleculeValue[{mol, mol2}, {"TorsionAngle", {{6, 1, 2, 3}, {1, 2, 3, 4}, {2, 3, 4, 5}}}]MoleculePlot3D /@ {mol, mol2}"SetUnpairedElectronCount" (1)
"TransformAtomCoordinates" (2)
Create a copy of a molecule with the coordinates translated in space:
m1 = Molecule["(2S)-3-hydroxy-2-[[(2S)-pyrrolidine-2-carbonyl]amino]propanoic acid"];m2 = MoleculeModify[m1, {"TransformAtomCoordinates", TranslationTransform[{Quantity[10, "Angstroms"], Quantity[10, "Angstroms"], Quantity[10, "Angstroms"]}]}]Show[MoleculePlot3D[#]& /@ {m1, m2}]Create a molecule's enantiomer using ReflectionTransform:
m = Molecule["(2S)-3-phenyl-2-[(2-phenylacetyl)amino]propanoic acid"]AtomList[m, Atom["AtomChirality" -> Except[None]], {"AtomIndex", "AtomChirality"}]m2 = MoleculeModify[m, {"TransformAtomCoordinates", ReflectionTransform[{0, 0, 1}, {0, 0, 4}]}]AtomList[m2, Atom["AtomChirality" -> Except[None]], {"AtomIndex", "AtomChirality"}]Show[MoleculePlot3D /@ {m, m2}, ViewPoint -> Front]Generalizations & Extensions (4)
"InvertAtom" (4)
mol = Molecule[{"C", "N", "C", "C", "C", Atom["C", "HydrogenCount" -> 1], "Cl", "C"},
{Bond[{1, 2}, "Single"], Bond[{2, 3}, "Single"], Bond[{3, 4}, "Single"], Bond[{2, 5}, "Single"],
Bond[{5, 6}, "Single"], Bond[{6, 7}, "Single"], Bond[{6, 8}, "Single"]},
{StereochemistryElements -> {Association["StereoType" -> "Tetrahedral", "ChiralCenter" -> 6,
"Direction" -> "Counterclockwise"]}}];
MoleculePlot3D[%, IconizedObject[«viewOptions»]]invMol = MoleculeModify[mol, {"InvertAroundAtom", 2}];
MoleculePlot3D[%, IconizedObject[«viewOptions»]]MoleculeValue[{mol, invMol}, {"OutOfPlaneAngle", {1, 2, 3, 5}}]Check that the chirality at the chlorine-bearing carbon has not changed:
MoleculeValue[{mol, invMol}, {"AtomChirality", 6}]Spiro centers can be inverted:
mol = Molecule[{"C", Atom["C", "HydrogenCount" -> 1], "O", "C", "C", "C", "C",
Atom["C", "HydrogenCount" -> 1], "Cl", "C", "C"}, {Bond[{1, 2}, "Single"],
Bond[{2, 3}, "Single"], Bond[{2, 4}, "Single"], Bond[{4, 5}, "Single"], Bond[{5, 6}, "Single"] ... "Direction" -> "Clockwise"], Association["StereoType" -> "Tetrahedral", "ChiralCenter" -> 2,
"Direction" -> "Counterclockwise"], Association["StereoType" -> "Tetrahedral",
"ChiralCenter" -> 8, "Direction" -> "Counterclockwise"]}}];
MoleculePlot3D[%, {1}, IconizedObject[«viewOptions»]]invMol = MoleculeModify[mol, {"InvertAroundAtom", 1}];
MoleculePlot3D[%, {1}, IconizedObject[«viewOptions»]]Check the chirality at the spiro atom:
MoleculeValue[{mol, invMol}, {"AtomChirality", 1}]Check that the chirality has not changed elsewhere:
MoleculeValue[{mol, invMol}, {"AtomChirality", {2, 8}}]Ring fusion atoms with sufficiently large rings can be inverted:
mol = Molecule[{"C", "C", "C", "C", "C", "C", Atom["C", "HydrogenCount" -> 1], "C", "C", "C", "C"},
{Bond[{1, 2}, "Single"], Bond[{1, 3}, "Single"], Bond[{3, 4}, "Single"], Bond[{4, 5}, "Single"],
Bond[{5, 6}, "Single"], Bond[{6, 7}, "Single"], Bond ... gle"]}, {StereochemistryElements ->
{Association["StereoType" -> "Tetrahedral", "ChiralCenter" -> 1,
"Direction" -> "Counterclockwise"], Association["StereoType" -> "Tetrahedral",
"ChiralCenter" -> 7, "Direction" -> "Clockwise"]}}];
MoleculePlot3D[%, {1}, IconizedObject[«viewOptions»]]invMol = MoleculeModify[mol, {"InvertAroundAtom", 1}];
MoleculePlot3D[%, {1}, IconizedObject[«viewOptions»]]MoleculeValue[{mol, invMol}, {"AtomChirality", 1}]Bridgehead atoms with sufficiently large rings can be inverted:
mol = Molecule[{"C", "C", "C", "C", "C", "C", "C", "C", "C", "C", "C"},
{Bond[{1, 2}, "Single"], Bond[{2, 3}, "Single"], Bond[{3, 4}, "Single"], Bond[{4, 5}, "Single"],
Bond[{5, 6}, "Single"], Bond[{6, 7}, "Single"], Bond[{7, 8}, "Single"], Bond[{5, 9}, "Single"],
Bond[{9, 10}, "Single"], Bond[{10, 11}, "Single"], Bond[{8, 1}, "Single"],
Bond[{11, 1}, "Single"]}, {}];
MoleculePlot3D[%, {1}, IconizedObject[«viewOptions»]]invMol = MoleculeModify[mol, {"InvertAroundAtom", 1}];
MoleculePlot3D[%, {1}, IconizedObject[«viewOptions»]]Applications (1)
"InvertAroundAtom" (1)
Invert the anomeric carbon of α-D-glucose:
alphaDglucose = Molecule["alpha-D-glucose"];
MoleculePlot3D[%, IconizedObject[«viewOptions»]]betaDglucose = MoleculeModify[alphaDglucose, {"InvertAroundAtom", 5}];
MoleculePlot3D[%, IconizedObject[«viewOptions»]]Check that the stereochemistry at only atom 5 has changed:
(AssociationThread[Range[#["AtomCount"]], MoleculeValue[#, "AtomChirality"]]//DeleteCases[None])& /@ {alphaDglucose, betaDglucose}Possible Issues (4)
"InvertAroundAtom" (4)
Ring fusion atoms with small rings are not inverted:
mol = Molecule[{"C", "C", "C", "C", "C", Atom["C", "HydrogenCount" -> 1], "C", "C", "C"},
{Bond[{1, 2}, "Single"], Bond[{1, 3}, "Single"], Bond[{3, 4}, "Single"], Bond[{4, 5}, "Single"],
Bond[{5, 6}, "Single"], Bond[{6, 7}, "Single"], Bond[{7, 8}, " ... ingle"]},
{StereochemistryElements -> {Association["StereoType" -> "Tetrahedral", "ChiralCenter" -> 1,
"Direction" -> "Counterclockwise"], Association["StereoType" -> "Tetrahedral",
"ChiralCenter" -> 6, "Direction" -> "Clockwise"]}}];
MoleculePlot3D[%, {1}]MoleculeModify[mol, {"InvertAroundAtom", 1}]MoleculeModify[Molecule[{"C", "C", "C", "C", "C", Atom["C", "HydrogenCount" -> 1], "C", "C", "C"},
{Bond[{1, 2}, "Single"], Bond[{1, 3}, "Single"], Bond[{3, 4}, "Single"], Bond[{4, 5}, "Single"],
Bond[{5, 6}, "Single"], Bond[{6, 7}, "Single"], Bond[{7, 8}, " ... ingle"]},
{StereochemistryElements -> {Association["StereoType" -> "Tetrahedral", "ChiralCenter" -> 1,
"Direction" -> "Counterclockwise"], Association["StereoType" -> "Tetrahedral",
"ChiralCenter" -> 6, "Direction" -> "Clockwise"]}}], {"InvertAroundAtom", 1}]Bridgehead atoms with small rings are not inverted:
mol = Molecule[{"C", "C", "O", "C", "C", "C", "C"}, {Bond[{1, 2}, "Single"], Bond[{2, 3}, "Single"],
Bond[{3, 4}, "Single"], Bond[{4, 5}, "Single"], Bond[{4, 6}, "Single"], Bond[{6, 7}, "Single"],
Bond[{5, 1}, "Single"], Bond[{7, 1}, "Single"]}, {}];
MoleculePlot3D[%, {1}, IconizedObject[«viewOptions»]]MoleculeModify[mol, {"InvertAroundAtom", 1}]Bridgehead atoms in tricyclic and larger ring systems are not inverted:
mol = Molecule[{"C", "C", "C", "C", "C", "C", "C", "C", "C", "C", "C"},
{Bond[{1, 2}, "Single"], Bond[{2, 3}, "Single"], Bond[{3, 4}, "Single"], Bond[{4, 5}, "Single"],
Bond[{5, 6}, "Single"], Bond[{6, 7}, "Single"], Bond[{7, 8}, "Single"], Bond[{5, 9}, "Single"],
Bond[{9, 10}, "Single"], Bond[{10, 11}, "Single"], Bond[{5, 1}, "Single"],
Bond[{8, 1}, "Single"], Bond[{11, 1}, "Single"]}, {}];
MoleculePlot3D[%, {1}, PlotTheme -> "Tubes", IncludeHydrogens -> None, IconizedObject[«viewOptions»]]MoleculeModify[mol, {"InvertAroundAtom", 1}]mol = Molecule[{"C", "C", "C", "C", "C", "C", "C", "C", "C", "C", "C", "C", "C", "C", "C", "C", "C"},
{Bond[{1, 2}, "Single"], Bond[{2, 3}, "Single"], Bond[{3, 4}, "Single"], Bond[{4, 5}, "Single"],
Bond[{2, 6}, "Single"], Bond[{6, 7}, "Single"], Bo ... e"],
Bond[{1, 13}, "Single"], Bond[{13, 14}, "Single"], Bond[{14, 15}, "Single"],
Bond[{15, 16}, "Single"], Bond[{16, 17}, "Single"], Bond[{9, 1}, "Single"],
Bond[{17, 1}, "Single"], Bond[{13, 5}, "Single"], Bond[{17, 12}, "Single"]}, {}];
MoleculePlot3D[%, {1}, PlotTheme -> "Tubes", IncludeHydrogens -> None]MoleculeModify[mol, {"InvertAroundAtom", 1}]Pentacoordinate atoms cannot be inverted:
mol = Molecule[{"P", "C", "C", "C", "C", "O", "C", "C", "F", "F", "F", "C", "C", "N", "C", "C", "C", "C",
"Cl"}, {Bond[{1, 2}, "Single"], Bond[{2, 3}, "Single"], Bond[{2, 4}, "Single"],
Bond[{2, 5}, "Single"], Bond[{1, 6}, "Single"], Bond[{6, 7}, " ... "],
Bond[{7, 12}, "Single"], Bond[{12, 13}, "Single"], Bond[{1, 14}, "Single"],
Bond[{14, 15}, "Single"], Bond[{15, 16}, "Single"], Bond[{14, 17}, "Single"],
Bond[{17, 18}, "Single"], Bond[{1, 19}, "Single"], Bond[{13, 1}, "Single"]}, {}];
MoleculePlot3D[%, PlotTheme -> "Tubes", IncludeHydrogens -> None, IconizedObject[«viewOptions»]]MoleculeModify[mol, {"InvertAroundAtom", 1}]Neat Examples (1)
Add distance constraints to the MMFF force field to find a conformation with a given distance between two atoms. First, create the base molecule:
m = Molecule[{Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"],
Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["C"],
Atom["C"], Atom["C"], Atom["C"], Atom["C"], Atom["O"], A ... r" -> 33,
"Direction" -> "Clockwise", "FiducialAtom" -> 32, "Ligands" -> {34, 35, 107}],
Association["StereoType" -> "Tetrahedral", "ChiralCenter" -> 38, "Direction" -> "Clockwise",
"FiducialAtom" -> 37, "Ligands" -> {39, 40, 117}]}];Find the two atoms that are farthest away in the standard configuration:
dmat = m["DistanceMatrix"]{a1, a2} = FirstPosition[Normal[dmat], Max[dmat]]The default conformation has these two atoms 32 ångströms apart:
dmat[[a1, a2]]MoleculePlot3D[m, {Style[a1, Blue], Style[a2, Red]}]Use "EnergyMinimizeAtomCoordinates" to find new conformations with specific separations for these atoms:
mod = MoleculeModify[m, {"EnergyMinimizeAtomCoordinates", {a1, a2} -> #}]& /@ {Quantity[5, "Angstroms"], Quantity[10, "Angstroms"], Quantity[20, "Angstroms"], Quantity[40, "Angstroms"]}Verify that the returned geometries match the requirement and get the force field energies:
MoleculeValue[mod, {{"InteratomicDistance", {a1, a2}}, "MMFFEnergy"}]MoleculePlot3D /@ modText
Wolfram Research (2019), MoleculeModify, Wolfram Language function, https://reference.wolfram.com/language/ref/MoleculeModify.html (updated 2026).
CMS
Wolfram Language. 2019. "MoleculeModify." Wolfram Language & System Documentation Center. Wolfram Research. Last Modified 2026. https://reference.wolfram.com/language/ref/MoleculeModify.html.
APA
Wolfram Language. (2019). MoleculeModify. Wolfram Language & System Documentation Center. Retrieved from https://reference.wolfram.com/language/ref/MoleculeModify.html