



MoleculeModify
MoleculeModify[mol,"mod"]
给出对分子 mol 应用修改 "mod" 后得到的分子或分子列表.
MoleculeModify[mol,{"mod",specs}]
给出对分子 mol 应用带有额外指定 specs 的修改 "mod" 后得到的分子或分子列表.
MoleculeModify["mod"]
表示 MoleculeModify 的算符形式,可应用于分子.
更多信息和选项











- 修改分子的原子列表的操作:
-
{"AddAtom",atom} 添加不相连的原子 {"AddAtom",{atom1,atom2,…}} 添加一系列原子 {"DeleteAtom",a} 删除索引为 a 的原子 {"DeleteAtom",{a1,a2,…}} 删除列出的原子 {"ReplaceAtom",aatom} 将索引为 a 的原子替换为 atom {"ReplaceAtom",{a1atom1,…}} 用 atomi 替换原子 ai - atom 应为以下形式:
-
"sym" 原子符号为 "sym"(如 "C"、"Cl")的原子 Entity["Element",element] 给定元素的原子 Entity["Isotope",isotope] 指定同位素的原子 Atom["sym","prop"val,…] 具有指定属性的原子 - "AddAtom" 将添加一个不相连的原子,与分子的其余部分之间没有键. 如果要添加一个新的原子和键,请使用 "AddBond".
- "DeleteAtom" 将删除该原子的所有键.
- 当去除或替换一个原子时,可以调整显式氢原子的数量以保持合理的化合价. 断开的氢原子将会被删除. 如果想禁用此行为,请使用选项 ValenceErrorHandlingFalse.
- 按正常方式将隐式氢原子放入添加的原子中. 如果想禁用隐式氢原子,在新原子指定中使用 "HydrogenCount"None.
- 修改原子属性的操作有:
-
{"SetFormalCharge",aval} 将索引为 a 的原子的形式电荷设为 val {"SetMassNumber",aval} 将索引为 a 的原子的质量数设为 val {"SetUnpairedElectronCount",aval} 为给定原子设置游离电子数 - 如果想改变多个原子的属性,可使用规则列表 {a1val1,a2val2,…}.
- 修改分子的键列表的操作:
-
{"AddBond",{a1,a2}} 在索引为 a1 和 a2 的原子间添加单键 {"AddBond",Bond[{a1,a2},"type"]} 在给定原子间添加给定类型的键 {"AddBond",Bond[{a1,atom},type]} 索引为 a1 的原子和由 atom 表示的新原子之间的键 {"DeleteBond",{a1,a2}} 移除给定索引的原子间的键 {"SetBondType",{a1,a2}"type"} 设置索引为 a1 和 a2 的原子间的键的类型 -
"SetAromaticity" 将局部单键和双键转换为芳香键 "Kekulize" 将芳香键转换为局部单键和双键 {"RenumberAtoms",{a1,a2,…}} 对原子列表重新排序,使得索引为 ai 的原子出现在原子列表中的位置 i {"SetMetaInformation",meta} 将分子的 MetaInformation 设为 meta,给定应为一个关联或键-值对 "MakeHydrogensExplicit" 将隐式氢原子转换为显式氢原子 "MakeHydrogensImplicit" 当从正常价态规则可以推断出氢原子的存在时,去除显式氢原子 - 关于分子的三维排列的信息(与三维几何结构无关)被保存在 StereochemistryElements 选项值中. 以下操作将改变 立体元素:
-
"AssignStereochemistryFromCoordinates" 根据 3D 坐标分配四面体和双键立体化学元素 "RemoveStereochemistry" 删除任何已定义的立体化学信息 - 如果要修改原子或键的已定义的立体化学信息,请使用以下操作:
-
{"SetAtomChirality",aval} 设置原子的手性值为 "R" 或 "S" {"SetBondStereo",{a1,a2}val} 将键的立体值设为 "E" 或 "Z" - 修改原子或键的立体结构将影响按要求生成的 3D 坐标. 如果分子已经包含有 3D 坐标,在修改立体化学信息时会将其丢弃.
-
{"ReplaceSubstructure",pattrepl} 将所有的 patt 替换为 repl {"ReplaceSubstructure",pattrepl,attchmts} 用 repl 替换 patt,只有指定的附着点 {"ReplaceSubstructureList",pattrepl} 将所有的 patt 替换为 repl,并返回一个列表 - 此处,repl 应为 Molecule 对象或 SMILES 字符串,patt 可为以下形式:
-
MoleculePattern[…] 有效的分子模式对象 {a1,a2,…} 构成相连子结构的原子索引列表 {{a1,a2,…},{b1,b2,…},…} 原子索引列表组成的列表,将每个原子索引列表替换为 repl - attchmts 应为规则列表 {p1r1,…},从模式原子的索引到要替换的原子的索引. 如果没有提供附着点,则匹配模式和替换中的第一个和最后一个原子.
- 在二维或三维空间绘制分子时,分别从 AtomCoordinates 和 AtomDiagramCoordinates 选项值中获取坐标. 如果分子表达式中没有出现坐标,则自动生成坐标.
- 以下操作将预先自动计算坐标,并返回含有该坐标的 Molecule 对象:
-
"ComputeAtomCoordinates" 自动计算分子的三维坐标 {"ComputeAtomCoordinates",opts} 使用选项 opts 计算原子的坐标 {"GenerateConformers",n} 用不同的种子计算三维坐标 n 次,返回一系列分子 "ComputeAtomDiagramCoordinates" 计算二维坐标 - "ComputeAtomCoordinates" 将使所有隐式氢变为显式氢以便嵌入分子.
- "ComputeAtomCoordinates" 接受以下选项:
-
"Canonicalize" True 是否规范化坐标 "EnergyMinimize" True 是否在坐标上运行力场最小化 RandomSeeding 1234 怎样对随机性进行播种 - 默认情况下,"GenerateConformers" 将返回未被最小化的异象体.
- "ComputeAtomDiagramCoordinates" 将使与碳相连的氢变为隐式氢,使与杂原子相连的氢变为显式氢,以便与 MoleculePlot 的默认输出匹配. 通过使用 {"ComputeAtomDiagramCoordinates",IncludeHydrogensTrue} 可包含所有原子.
- 如果要修改单个原子的坐标,可使用:
-
{"SetAtomPosition",a{x,y,z}} 设置指定原子的位置 - 如果要规范化原子的坐标而不改变内部的几何结果,请使用:
-
"CanonicalizeAtomCoordinates" 将分子的质心平移到原点,并使主轴与直角坐标轴对齐 - 要翻转特定原子周围的局部几何结构,请使用:
-
{"InvertAroundAtom",a} 翻转索引为 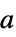 的原子周围的几何结构
的原子周围的几何结构{"InvertAroundAtom",{a1,a2,…}} 翻转多个原子周围的几何结构 - 四面体原子通过交换两个最小的可移动配体实现构型翻转. 三角锥形原子通过移动最小的可移动配体实现翻转.可移动原子要么不在环中,要么位于同一环内. 被移动原子的坐标通过二重旋转进行变换,因此分子中所有其他的立体中心保持不变.
- 对于普通的几何转换,可使用:
-
{"TransformAtomCoordinates",tfun} 对坐标应用转换函数 tfun - 此处,tfun 应为 TransformationFunction 对象,如 TranslationTransform、ReflectionTransform 等,或操作并返回 QuantityArray 对象的任意函数.
- 以下操作将修改内部坐标:
-
{"SetBondLength", {a1,a2}val} 设置指定原子之间的键长 {"SetBondAngle", {a1,a2,a3}val} 设置指定原子之间的角度 {"SetTorsionAngle",{a1,a2,a3,a4}val} 设置四个连续的由化学键联接的原子的扭转角 - 这些操作也支持长度列表或角度变化,如 {{a1,a2}val1,…}.
- 此处,val 应为 Quantity 对象或数字. 如果是数字,则假定单位为 "Angstroms" 或 "AngularDegrees".
- 对于 "SetBondLength",原子 a1 和 a2 必须成键,且它们之间的键不应成环. 与原子 a2 相连的所有原子都将被移除.
- 对于 "SetBondAngle",原子 {a1,a2} 和 {a2,a3} 应成键,且这两个键都不应成环. 与原子 a3 相连的所有原子都将被移除.
- 对于 "SetTorsionAngle",原子 {a1,a2}、{a2,a3} 和 {a3,a4} 应成键,且键 {a2,a3} 不应成环. 与原子 a4 相连的所有原子都将被移除.
- 如果要最小化相对于 MMFF 力场的坐标,请使用:
-
"EnergyMinimizeAtomCoordinates" 最小化总力场能量 {"EnergyMinimizeAtomCoordinates",ff} 用力场 ff("UFF"、"MMFF94" 或 "MMFF94s" 之一最小化能量) {"EnergyMinimizeAtomCoordinates",constraints} 在给定的力场约束条件下将能量最小化 - constraints 应为一系列键、角度或扭转角限制条件,可采用以下形式:
-
{a1,a2}d 指定两个原子间的距离 {a1,a2,a3}θ 指定三个原子间的角度 {a1,a2,a3,a4}θ 指定四个原子间的扭转角 <…> 一个关联,其键为 "Atoms"、"Value" 和 "ForceConstant"
对原子进行修改
对键进行修改
普通修改
对立体化学信息进行修改
替换子结构
生成坐标
对几何结构进行修改
范例
打开所有单元 关闭所有单元基本范例 (4)
范围 (40)
"CanonicalizeAtomCoordinates" (1)
从 "XYZ" 文件导入一个结构,并规范化三维坐标:
"CanonicalizeAtomCoordinates" 将平移分子,将质心移动到原点,然后旋转以使主轴与直角坐标轴对齐:
"ComputeAtomCoordinates" (2)
当使用诸如 SMILES 字符串或名称之类的标识符创建分子时,不会立即计算坐标:
用 "ComputeAtomCoordinates" 计算坐标并将其保存在分子表达式中:
使用 RandomSeeding 选项生成不同的分子构象:
"ComputeAtomDiagramCoordinates" (2)
当用 MoleculePlot 在二维空间可视化分子时,默认情况下,将与碳原子相连的氢原子隐藏起来. 默认情况下,在辛醇中只显示羟基氢:
"DeleteAtom" (2)
找出匹配的 Entity:
删除原子时,与其相连的所有氢也将被去除,用隐式氢填充重原子上未填充的化合价. 通过 ValenceErrorHandling->False 可禁用此行为,留下未配对的电子:
"EnergyMinimizeAtomCoordinates" (1)
"GenerateConformers" (1)
"InvertAroundAtom" (3)
"SetBondType" (1)
"SetMassNumber" (1)
"SetMetaInformation" (1)
"TransformAtomCoordinates" (2)
推广和延伸 (4)
文本
Wolfram Research (2019),MoleculeModify,Wolfram 语言函数,https://reference.wolfram.com/language/ref/MoleculeModify.html (更新于 2020 年).
CMS
Wolfram 语言. 2019. "MoleculeModify." Wolfram 语言与系统参考资料中心. Wolfram Research. 最新版本 2020. https://reference.wolfram.com/language/ref/MoleculeModify.html.
APA
Wolfram 语言. (2019). MoleculeModify. Wolfram 语言与系统参考资料中心. 追溯自 https://reference.wolfram.com/language/ref/MoleculeModify.html 年